Research
Research
多様な活性分子構造の生成
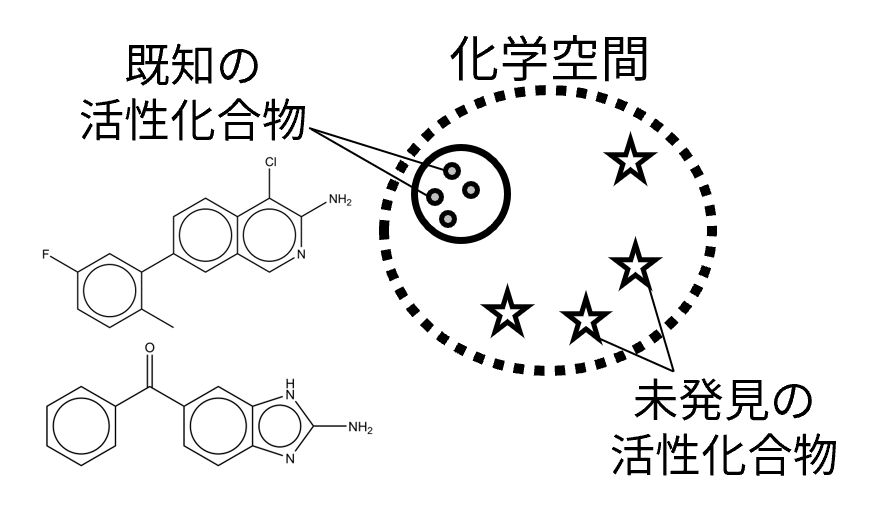
潜在的なドラッグライクな分子が存在する化学空間の大きさは 10の23~60乗程度と推定されていますが、これまで人間が合成した化合物の数は10の8乗程度であり[1,2]、多様な活性分子を効率的に探索する手法が求められています。現在、深層学習などの情報科学技術の急速な発展が創薬分野に大きな影響を与えています。中でも分子生成モデルは急速に発展している分野であり、データベースに含まれる分子に類似する新規分子構造の生成が可能となりました[3]。一方で分子生成モデルでは生成される分子構造の多様性が限定的であり、多様な分子構造を生成すると活性分子になる確率は低下すると考えられます。本研究では、所望の分子特性(物性など)が与えられた時に指定した特性値を満たしつつ、標的タンパク質に対して活性を示す多様な新奇分子を生成する手法を開発しています。
- Polishchuk PG, et al., Aided. Mol. Des. 2013;27(8):675–679.
- Reymond J-L, et al., ACS Chem Nerosci. 2012; 3:649–57.
- M. H. S. Segler et al., ACS Cent. Sci. 2018, 4, 1, 120–131.
合成展開性の定量化
低分子創薬では、活性発現に重要な構造を持つリード化合物に対して様々な官能基修飾や構造変換を行うことで、より薬として望ましい性質を持つように化合物を最適化します。しかし、リード化合物の中には修飾や変換自体が困難な化合物が存在します。そのため、合成展開性の高い化合物選択は、最適化の成否を左右する重要な要素です。現在、合成可能性や構造の複雑性などを表現する様々なスコアが開発されていますが、合成展開性を示す指標は存在せず、合成展開性の高い化合物の選択は困難です。そこで本研究では、合成展開性を評価する指標の開発に取り組んでいます。
深層学習を用いた化学反応の収率予測
化学反応における収率は、反応経路を決定するための判断基準となります。先例のない反応では、化学者は勘と経験をもとに反応条件を決定する。化学反応の収率予測が可能となれば、高い収率が期待される反応に焦点を当てることができます。そのため、合理的な反応経路の設計につながり、金銭的・時間的コストの削減に繋がります。大規模データに適用可能な機械学習を利用した収率予測の研究[1]では、計算コストの高い量子化学計算が必要であることに加えて、モデル構築に利用していない化合物を含む反応の収率予測精度が低いという課題があります。そこで、本研究では深層学習を用いてこの課題に取り組んでいます。
- D. T. Ahneman, J. G. Estrada, S. Lin, S. D. Dreher, A. G. Doyle. Science. 2018, 360, 186-190
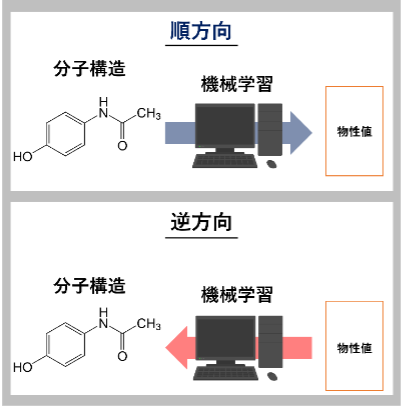
所望の物性を有する検証容易な分子設計手法の開発
所望の物性を有する分子構造の生成には,Inverse-QSPRを始めとする逆解析手法を用いた手法等があります。しかし、合成可能性を考慮していない、莫大な数に及ぶ分子構造のスクリーニングに対する計算コストが非常に大きいなどの欠点が挙げられます[1]。そのため、仮想分子を設計し、計算機で所望の物性を有するか検証した研究は多くありますが、実際の分子が所望の物性を有するかどうかを検証した研究は非常に少ないです。本研究では,所望の物性を有する目的分子の設計だけでなく、検証が容易な分子の設計手法を開発しています。また、共同研究先と協力し、現実世界においても設計した分子が所望の物性を有するのか検証します。
- Kaneko, et al.; JCAC 2015, 16, 15-29
ポテンシャルファーマコフォアポイントの再定義
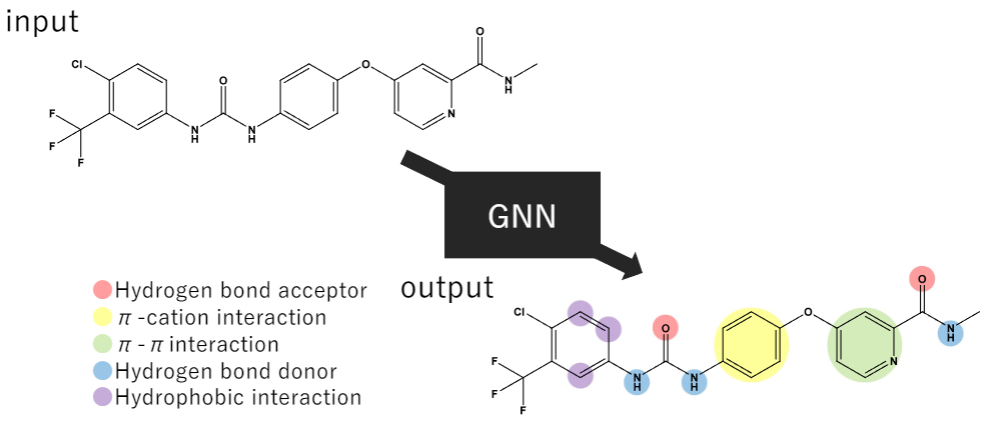
薬となる低分子と標的マクロ分子との分子間相互作用、および相互作用を形成する部分構造や原子であるポテンシャルファーマコフォアポイント (PPP)を理解することは、合理的な分子設計において重要です。従来の手法は、事前定義されたルールに則ってPPPを定めますが、実際の相互作用に基づいたPPPの定義が困難であるという課題があります。そこで本研究では、標的マクロ分子とリガンド複合体から得た相互作用情報を基に、PPPを予測するモデル開発に取り組んでいます
- Definitions for 2D Pharmacophores from: Gobbi and Poppinger, Biotech. Bioeng. _61_ 47-54 (1998)
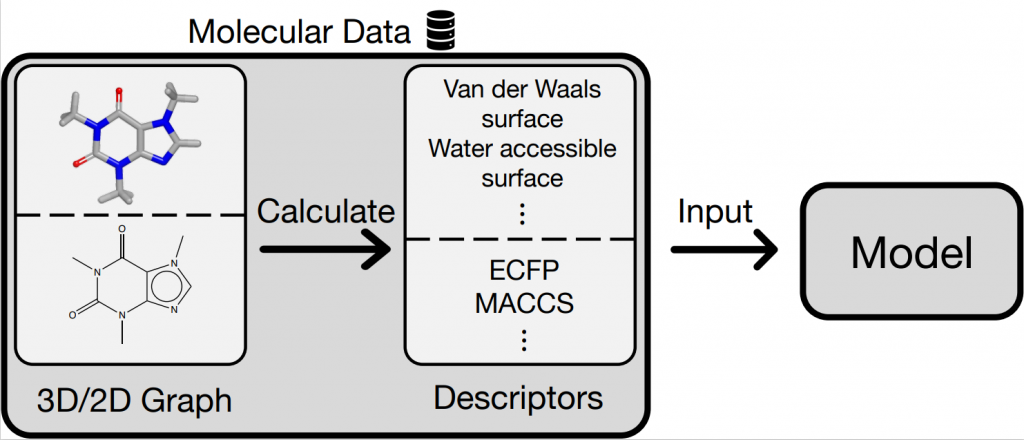
適切な記述子の自動選択可能なモデルの開発
ケモインフォマティクスにおいて、分子の物性・活性を正確かつ高速に予測することは重要な課題です。機械学習モデルによる学習・予測には化合物の数値表現が必要です。その表現方法は大きく分けて、2次元(回転可能な結合数など)と3次元(分子の表面積など)表現があります。しかし、どちらがより優れているかは解明されておらず[1]、予測対象によって異なる場合も多々あります。そこで本研究では、予測対象に応じて2次元表現と3次元表現を用いるか自動的に選択するモデルの開発を行っています。
[1] Gao, Kaifu, et al. “Are 2D fingerprints still valuable for drug discovery?.” Physical chemistry chemical physics 22.16 (2020): 8373-8390.